|
Schizophrenia... Gray Matter Loss... With A Non-Genetic Trigger... The following is taken from an article that appeared on the National Institute of Mental Health website on November 8, 2001. Note the statement that a "non-genetic trigger" appears to be at play here...
The following is taken word for word from the Proceedings of the National Academy of Sciences: PNAS | September 25, 2001 | vol. 98 | no. 20 | 11650-11655, posted at: http://www.pnas.org/cgi/content/full/98/20/11650 ... but, just in case this "disappears"... here's the article... clicking on the pictures should make them "bigger/more easy to see". In article below, click on the link where the picture should be to bring up the picture!
From the Cover
|

| |
Abstract |
|---|
Neurodevelopmental models for the pathology of schizophrenia propose both polygenetic and environmental risks, as well as early (pre/perinatal) and late (usually adolescent) developmental brain abnormalities. With the use of brain mapping algorithms, we detected striking anatomical profiles of accelerated gray matter loss in very early-onset schizophrenia; surprisingly, deficits moved in a dynamic pattern, enveloping increasing amounts of cortex throughout adolescence. Early-onset patients were rescanned prospectively with MRI, at 2-year intervals at three time points, to uncover the dynamics and timing of disease progression during adolescence. The earliest deficits were found in parietal brain regions, supporting visuospatial and associative thinking, where adult deficits are known to be mediated by environmental (nongenetic) factors. Over 5 years, these deficits progressed anteriorly into temporal lobes, engulfing sensorimotor and dorsolateral prefrontal cortices, and frontal eye fields. These emerging patterns correlated with psychotic symptom severity and mirrored the neuromotor, auditory, visual search, and frontal executive impairments in the disease. In temporal regions, gray matter loss was completely absent early in the disease but became pervasive later. Only the latest changes included dorsolateral prefrontal cortex and superior temporal gyri, deficit regions found consistently in adult studies. These emerging dynamic patterns were (i) controlled for medication and IQ effects, (ii) replicated in independent groups of males and females, and (iii) charted in individuals and groups. The resulting mapping strategy reveals a shifting pattern of tissue loss in schizophrenia. Aspects of the anatomy and dynamics of disease are uncovered, in a changing profile that implicates genetic and nongenetic patterns of deficits.
| |
Introduction |
|---|
Little is known about the profile of brain change in adolescence and its modulation in diseases with adolescent onset. Schizophrenia, for example, has typical onset in late adolescence or early adulthood. Cases occurring in childhood or early adolescence, however, present unique opportunities to study disease development during adolescence. Childhood-onset schizophrenia (COS) is a severe form of the disorder that appears to be clinically and neurobiologically continuous with the later onset illness (1). The causes of schizophrenia are not known, but it is increasingly considered a neurodevelopmental disorder (2, 3). Both early (prenatal) and later abnormalities of brain development have been proposed (4-6). However, neither the anatomical pattern nor the timing of these developmental events has been established.
In response to these challenges, we designed a brain mapping strategy to uncover deficit patterns as they emerged in populations imaged longitudinally through adolescence for 5 years. Because gray matter loss is implicated in schizophrenia and is also known to occur in adolescence (7-15), we set out to create detailed spatiotemporal maps of these loss processes. Their timing and anatomical profile are fundamental to understanding how the disease emerges; so far it has been difficult to test hypotheses about genetic and environmental triggers of schizophrenia because the topography and dynamics of the disease, especially at the cortex, are not well understood. In a recent cross-sectional genetic study based on a cohort of 80 adult twins discordant for schizophrenia,§ we isolated a genetic continuum in which cortical deficits were found in gradually increasing degrees, in individuals with increasing genetic affinity to a patient. By controlling for common genotype, we isolated discrete regions of cortex whose deficits were attributable to genetic and to nongenetic factors, although the emergence and timing of these deficits could not be evaluated.
The current study was intended to chart the emergence of these deficits in a severely affected cohort followed for 5 years, revealing an unsuspected developmental trajectory in these schizophrenic adolescents. This technique uncovered a dynamic wave of accelerated gray matter loss, spreading from parietal cortices at disease onset to encompass temporal and frontal regions later in the disease. The rates and temporal sequencing of cortical gray matter loss was mapped in the teenage years and was found to be greatly accelerated in diseased relative to healthy teenagers matched for age, gender, and demographics. The final profile was consistent with the loss pattern in adult schizophrenia. We also correlated loss rates with symptom severity and controlled for potential medication and IQ effects. Local changes were examined in relation to genetic and nongenetic deficit patterns found in adults. This study is therefore a three-dimensional visualization of the timing, rates, and anatomical distribution of brain structure changes in adolescents with schizophrenia. It suggests a dynamic structural basis for early prodromal symptoms and for the positive and negative deficit symptoms observed clinically (16).
| |
Methods |
|---|
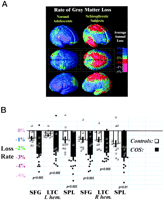
Summary. Three-dimensional maps of brain change were derived from high-resolution magnetic resonance images (MRI scans) acquired repeatedly from the same subjects over a 5-year time span. Twelve schizophrenic subjects (aged 13.9 ± 0.8 years at first scan) and a parallel group of 12 healthy adolescents (aged 13.5 ± 0.7 years at initial scan) were imaged repeatedly for 4.6 years (the combined groups were scanned every 2.3 years ± 1.4 months (SD); for clarity this is referred to as 5 years). Patients and controls were matched for age, gender, and demographics and were scanned identically on the same scanner at exactly the same ages and intervals. The three-dimensional distribution of gray matter in the brain was computed, as in previous studies of Alzheimer's disease (17), and was compared from one scan to the next with the use of a computational cortical pattern matching strategy that aligns corresponding locations on the cortical surface, across time and across subjects. This procedure allowed us to pool maps of individual gray matter loss over time. Average rates of gray matter loss were computed for each group and compared across corresponding regions of cortex (Fig. 1), before a more detailed analysis of nonlinear and age-dependent effects. The amount of loss and the rate of loss were separately evaluated. Findings were also examined in relation to genetic and nongenetic patterns found in recent studies of adult patients.
|
Subjects and Imaging. Subjects were recruited as part of an ongoing National Institute of Mental Health study of COS (1) and evaluated prospectively over a 5-year time period. Twelve patients (six males and six females) and 12 healthy volunteers (six males and six females), as well as an additional medication-matched group (see below), were followed longitudinally. All patients satisfied DSM-III-R diagnostic criteria for schizophrenia (18), with onset of psychotic symptoms by age 12. All patients had a history of poor response to, or intolerance of, at least two typical neuroleptics. They had a mean full-scale IQ at study entry of 70.4 ± 12.9 SD and no other active neurological or medical disease. Diagnosis was determined from clinical and structured interviews with the adolescents and their parents based on portions of the Schedule for Affective Disorders and Schizophrenia for School-Age Children-Epidemiologic Version (19) and of the Diagnostic Interview for Children and Adolescents Revised (20), as well as from previous records. Psychopathological symptoms were evaluated with the use of the Scales for the Assessment of Positive and Negative Symptoms (SAPS/SANS) (21) and the Brief Psychiatric Rating Scale (22) (see ref. 1 for details). Normal adolescent controls were screened for medical, neurologic, and psychiatric illness and learning disabilities as described (1). We rigorously matched the cohorts for age (see below), gender, follow-up interval (which was identical), social background, and height.
Medication and IQ-Matched Group. Medication effects were assessed by analyzing a second group of nonschizophrenic medication-matched subjects. The 10 age- and gender-matched psychosis patients not otherwise specified (NOS) (1, 23) received the same medication as the schizophrenic group at baseline and follow-up but did not satisfy DSM-III-R criteria for schizophrenia. They were also IQ-matched with the COS patients (mean IQ: 76 ± 10 SD) and matched for age, gender, and demographics with the healthy controls. These children had very transient psychotic symptoms, emotional lability, poor interpersonal skills, normal social interest, and multiple deficits in information processing (1). They were less severely impaired than the COS group but continued with a mixture of mood and behavior problems. None at follow-up was schizophrenic but rather exhibited chronic mood disturbance and lack of behavioral control; they were treated with neuroleptics for these symptoms (at doses similar to that used for COS; see below), which were quite effective in controlling these behaviors.
Of the 10 psychosis NOS patients, two patients received 300 and 450 mg clozapine (mean dose 375 mg/day), six received risperidone (2-8 mg; mean dose 5.25 ± 2.4 mg/day) [four in combination with valproic acid (mean 1,025 mg/day) and one in combination with olanzapine (20 mg/day)], and two were drug-free.
Magnetic Resonance Imaging. Three-dimensional (2562 × 124 resolution) T1-weighted fast SPGR (spoiled gradient) MRI volumes were acquired from all 34 subjects. All images were acquired with the same 1.5-T Signa scanner (General Electric) located at the National Institutes of Health Clinical Center (Bethesda, MD). Imaging parameters were as follows: time to echo, 5 ms; time to repeat, 24 ms; flip angle, 45°; and field of view, 24 cm. The same set of 12 healthy controls was scanned at baseline (aged 13.5 ± 0.7 years) and ultimately after a 5-year interval (mean interval: 4.6 ± 0.2 years; age: 18.0 ± 0.8 years). In parallel, the 12 age- and gender-matched schizophrenic subjects were identically scanned at the exact same ages and intervals (mean age at first scan: 13.9 ± 0.8 years; 18.6 ± 1.0 years at final scan; mean interval: 4.6 ± 0.3 years). All subjects (controls, schizophrenic subjects, and the medication controls) were scanned three times, first at baseline, then a mean of 2.3 years later, and then again 4.6 years later. The combined groups were scanned every 2.3 years ± 1.4 months (SD).
Image Processing and Analysis. For each scan pair, a radio-frequency bias field correction algorithm eliminated intensity drifts due to scanner field inhomogeneity. The initial scan was then rigidly aligned (registered) to the target (24) and a supervised tissue classifier generated detailed maps of gray matter, white matter, and cerebrospinal fluid (25). A nearest-neighbor tissue classifier then assigned each image voxel to a particular tissue class (gray, white, or cerebrospinal fluid) or to a background class (15). Gray matter maps were retained for subsequent analysis.
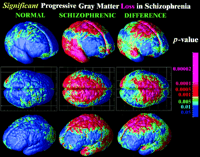
Three-Dimensional Cortical Maps. To compare and pool cortical data across subjects, a high-resolution surface model of the cortex was automatically extracted for each subject and time point. Based on the cortical models we created for each subject at different time points, a three-dimensional deformation vector field was computed that captured the shape change in the brain surface across the time interval. This method allows us to accommodate any brain shape changes when comparing cortical gray matter within a subject across time. Given that the deformation maps associate cortical locations with the same relation to the primary folding pattern across subjects, a local measurement of gray matter density was made in each subject and averaged across equivalent cortical locations. To quantify local gray matter, we used a measure termed gray matter density (15, 17, 26, 27). This method measures the proportion of gray matter in a small region of fixed radius (5 mm) around each cortical point. Given the large anatomic variability in some cortical regions, high-dimensional elastic matching of cortical patterns (17, 28) was used to associate measures of gray matter density from homologous cortical regions across subjects and across time. Annualized four-dimensional maps of gray matter loss rates within each subject were elastically realigned for averaging and comparison across diagnostic groups. Statistical maps were generated that indicated locally the degree to which gray matter loss rates were statistically linked with diagnosis, gender, and positive or negative symptoms (SAPS/SANS) (21). The P value describing the significance of this linkage was plotted at each point on the cortex with a color code to produce a statistical map (e.g., Fig. 2). A null distribution was developed for the area of the average cortex with statistics above a fixed threshold in the significance maps (17, 29), so the significance of the loss patterns could be assessed after the appropriate correction for multiple comparisons.
|
| |
Results |
|---|
In schizophrenic patients, a striking accelerated loss of gray matter (peak values > 5% loss/year; Fig. 1A) was observed in a broad anatomical region encompassing frontal eye fields and supplementary motor, sensorimotor, parietal, and temporal cortices in both brain hemispheres (see Fig. 1). Average loss rates were significantly faster in patients in superior parietal lobules (left hemisphere mean ± standard error: 2.9 ± 0.5%/year, right hemisphere: 2.9 ± 0.5%/year; in controls: 1.1 ± 0.4 and 1.4 ± 0.5; group difference: P < 0.005 and 0.01), in superior frontal cortices (L/R: 2.6 ± 0.5 and 2.7 ± 0.4 in patients; 0.9 ± 0.3 and 1.0 ± 0.3 in controls; group difference: P < 0.003 and 0.002), and in lateral temporal cortices (L/R: 2.3 ± 0.9 and 2.4 ± 0.4 in patients; 0.7 ± 0.2 and 1.1 ± 0.3 in controls; group difference: P < 0.003 and 0.005). Subtle but significant changes were detected in normal adolescents (0.9-1.4% average loss per year; all regions showed significant loss, at P < 0.02). The schizophrenia group exhibited a region of intense, severely progressive loss, terminating anteriorly in the frontal eye fields and encompassing the temporal cortices.
Significance of the Progressive Loss. To understand whether these changes could be normal fluctuations, the variability in both the anatomical distribution and loss rates for gray matter were computed locally across the cortex, and the significance of the changes was established. Again, schizophrenic subjects underwent a significant, pervasive, and unrelenting loss of gray matter (P < 0.00002, all P values corrected), with progressive deficits throughout superior frontal, motor, and parietal brain regions, and a separate loss pattern observed in temporal cortices. Normal adolescents also lost tissue (P < 0.05, in parietal regions) even after normal variability was accounted for (Fig. 2A). A subtraction map was created to emphasize the fundamental loss pattern specific to the disease (Fig. 2C). Regions of progressive loss, in both anterior frontal and temporal cortices, were anatomically circumscribed in both the percentage loss and significance maps and appeared to terminate anteriorly in the frontal eye fields (Figs. 1 and 2). These figures show regions where tissue loss is faster in diseased than in normal adolescents. The same anatomically specific, dynamic profiles of tissue loss were replicated in independent samples of male and female schizophrenic patients (Fig. 3), suggesting that a similar profile and degree of progressive gray matter loss may operate in schizophrenia, irrespective of gender.
|

Nonlinear Loss. We further hypothesized that loss rates would be relatively greater in younger patients, possibly reflecting a more severe neurodevelopmental abnormality that may have led to an earlier illness onset and/or a disease-related exaggeration of nonlinear normal developmental processes. Right parietal and sensorimotor cortices (Fig. 4) underwent significantly faster loss in the younger adolescents, consistent with recent findings of overall volume reductions specific to parietal lobes in younger patients (30). In other brain regions, the rates of gray matter loss were not strongly affected by age, corroborating the use of annual averages to describe the dynamic pattern. Although nonlinear effects in other brain regions may be detectable in a much larger cohort, similar annual rates of loss were observed consistently in subjects throughout our sample, and across independent samples (Fig. 3).
|

Early Deficits. Because of the apparent sparing of inferior frontal cortices in the dynamic maps (Figs. 1 and 2) and their appearance in our recent cross-sectional studies of adult schizophrenia,§ we were concerned that earlier (perinatal or prepubertal) nonprogressive maldevelopment may not have been observed in the dynamic maps, as these maps only capture loss that intensifies over time. To detect earlier loss, we compared gray matter profiles across all 24 subjects at their first scan (Fig. 5 Upper) and at their last scan 4.6 years later (Fig. 5 Lower). Two striking features emerged. First, the severe progressive lateral temporal and dorsolateral prefrontal cortex deficit observed later was not apparent at age 13, even at a mean of 3 years after the onset of psychotic symptoms. These deficits, which are characteristic of adult and childhood schizophrenia (1, 31), were severely progressive after illness onset (on average 5% lateral temporal attrition per year) but were absent in the early phase of the disease. In evaluating the power of the approach, we would have been likely to detect an early temporal and dorsolateral prefrontal cortex deficit, if present, but the near identity (to within 0-1%) of average normal and patient gray matter distribution at first scan, combined with the high rate of progression and significant deficit observed later (Fig. 5B), jointly suggest that the process of temporal and prefrontal gray matter attrition is a later developmental event. Second, parietal and motor cortices showed a severe early deficit (up to 20% loss; P < 0.0005) with diffuse loss in other (but not temporal) cortical regions. This early (prepubertal) parietal deficit is consistent with the faster parietal loss found in younger patients (Fig. 4), whereas the dynamics of loss in other regions is more uniform with age. The initial parietal deficit, which is also progressive (Fig. 1), also occurs in regions where normal adolescents lose gray matter, although in disease this loss process is significantly accelerated (Fig. 2). Although it is unclear whether the normal and aberrant processes have a similar mechanism or are independent, the parietal and motor cortical deficits are progressive and are the earliest to develop. We recently found that the parietal regions, specifically, are also in deficit in adult patients relative to genetically identical controls (their monozygotic discordant twins). This finding indicates that environmental and not purely genetic factors are implicated in triggering this deficit, at least in adults.§ [emphasis added]. In the present study, a dynamic wave of progression from parietal cortices occurs later, into superior frontal, dorsolateral prefrontal, and temporal cortices (including superior temporal gyri; Fig. 5). These regions comprise a specific band of cortical territory in which adult deficits are thought to be strongly influenced by genetic factors (32),§ as deficits here (i) are found in unaffected relatives and (ii) significantly covary with an individual's degree of genetic affinity to a patient. In our adolescent cohort, the temporal and dorsolateral prefrontal cortex deficits were among the most severe but began in late adolescence and were observed only after symptom onset (Fig. 5).
|

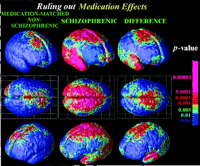
Medication-Matched Subjects. To address the possibility that neuroleptic exposure and/or lower IQ could have determined differential gray matter loss in the schizophrenics, we mapped 10 serially imaged subjects referred to the childhood schizophrenia study who did not meet diagnostic criteria for schizophrenia [labeled psychosis NOS, in DSM (18) terms] (23). These subjects received medication identical to that of the patients in this study through adolescence, primarily for control of aggressive outbursts, and at follow-up, none had progressed to schizophrenia (33) but all continued to exhibit chronic mood and behavior disturbance. Although medication is unlikely to be responsible for a loss profile that moves across the brain, clozapine, for example, may increase Fos immunoreactivity in the thalamus (34) and might, logically, modulate rates of cortical change. (In addition, brain regions important for motor function, including the basal ganglia, show increased volumes in response to some older, conventional neuroleptics, although these effects are renormalized after treatment with the atypical antipsychotics used in this study.) As seen in Fig. 6, although the nonschizophrenic group did show some subtle but significant tissue loss, this loss was much less marked than for the schizophrenics. Moreover, no temporal lobe deficits were observed in the psychosis NOS group (Fig. 6), suggesting that the wave of disease progression into temporal cortices may be specific to schizophrenia, regardless of medication and gender or IQ. Intriguingly, the psychosis NOS subjects, who share some of the deficit symptoms but do not satisfy criteria for schizophrenia, exhibited significantly accelerated gray matter loss in frontal cortices relative to healthy controls, in approximately the same (but a less pervasive) region as schizophrenics (a significant loss of 1.9% ± 0.7%/year was detected in both left and right superior frontal gyri; P < 0.03). Groups of healthy controls, psychosis NOS, and schizophrenic patients therefore lost frontal gray matter at successively increasing rates, i.e., in a statistical hierarchy with loss rates significantly faster in the nonschizophrenic control group than in healthy controls, and even faster in schizophrenia. In the region where the medication controls were affected (superior frontal cortices), their deficits at follow-up averaged 7.5% ± 1.6% relative to healthy controls (P < 0.006). This deficit was significantly less severe (P < 0.05) than the 13.0% ± 3.2% deficit in the schizophrenic group (P < 0.001, relative to healthy controls), whose global functioning was more greatly impaired at follow-up (P < 0.05; compare Children's Global Assessment of Functioning Scale scores, Table 1).
|
|
Relationship to Clinical Deficits. We further evaluated the clinical specificity and functional correlates of these findings. The patient group deteriorated overall; their Children's Global Assessment of Functioning Scale scores, which provide a global assessment of function, deteriorated from 37.4 ± 5.7 at study entry to 30.8 ± 3.4 at follow-up (P < 0.05; Table 1). Meanwhile, the average scores for the IQ/medication control group remained stable (43.0 ± 6.5 at entry; 41.9 ± 3.8 at follow-up) and were higher (less impaired) than those of the schizophrenic patients at follow-up (P < 0.05). This finding suggests an overall deterioration of global functioning in COS, consistent with the progressive deterioration of structure.
At an individual level, rates of temporal loss correlated strongly with a
SAPS total score at final scan (21)
(P < 0.015, left hemisphere; P < 0.004, right
hemisphere; all p values corrected). Faster loss in both
the superior temporal gyri and the entire temporal cortices was
significantly associated with a more severe clinical profile of
positive symptoms (e.g., hallucinations or delusions). Although
tissue loss rates were not significantly linked with the rate of
change in SAPS scores from baseline (P > 0.05), and SAPS
scores were not linked with the amount of tissue at baseline (P > 0.05),
loss rates were a good predictor of positive symptoms at
follow-up, i.e., the remaining symptoms that were refractory to
medication. In addition, those with the least overall tissue
deficit had the best cognitive performance in terms of full-scale
IQ at follow-up, and those with the worst deficit on MRI had the
lowest full-scale IQ at follow-up (r = 0.62; P < 0.016).
Gray matter quantity at initial scan was also a good predictor
of full-scale IQ in the patient group at follow-up (r = 0.52;
P < 0.042). At baseline, this linkage did not reach
significance (r = 0.44; P = 0.077), but a change in
correlations between baseline and follow-up was not significant (r2 ![]() r1 = 0.18;
z = 0.54; P = 0.3). Faster loss rates in frontal
cortex were also strongly correlated with more severe negative
symptoms (e.g., flat affect, poverty of speech; P < 0.038 for
total SANS score at final scan). This linkage is consistent with
the physiological hypothesis that negative symptoms of
schizophrenia may depend on reduced dopaminergic activity in
frontal cortices (35).
The tight linkage between the deficit symptoms of schizophrenia
and the pervasive loss of cortical tissue suggests a disease
mechanism that may only be partially opposed by neuroleptics (36).
r1 = 0.18;
z = 0.54; P = 0.3). Faster loss rates in frontal
cortex were also strongly correlated with more severe negative
symptoms (e.g., flat affect, poverty of speech; P < 0.038 for
total SANS score at final scan). This linkage is consistent with
the physiological hypothesis that negative symptoms of
schizophrenia may depend on reduced dopaminergic activity in
frontal cortices (35).
The tight linkage between the deficit symptoms of schizophrenia
and the pervasive loss of cortical tissue suggests a disease
mechanism that may only be partially opposed by neuroleptics (36).
| |
Discussion |
|---|
During the development of schizophrenia in these early adolescent subjects, a dynamic wave of gray matter loss occurred, starting in parietal association cortices and proceeding frontally to envelop dorsolateral prefrontal cortex and temporal cortices, including the superior temporal gyri. The deficits spread and intensified, in the same subjects, over 5 years of disease progression and eventually engulfed parietal, motor and supplementary motor, temporal (including primary auditory), and prefrontal cortices. The dynamic pattern is intriguing, as it begins in brain regions where deficits, at least in adults, appear to be mediated by environmental (nongenetic) factors (parietal cortices).§ It then progresses over a multiyear time frame into frontal and temporal regions where deficits appear, from our twin and other familial studies, to be strongly mediated by genetic factors (32).
Relation to Prior Findings. The dynamic pattern of loss may also suggest a structural basis for the prodromal and chronic neuromotor, sensory, and associative deficits observed clinically and in studies of the functional and metabolic integrity of the cortex. Glucose metabolism is reduced in frontal cortices in chronic childhood and adult schizophrenics both at rest and during the performance of tasks that increase frontal lobe metabolism, such as the Continuous Performance Test (37). COS patients also display significantly increased metabolic rates in inferior frontal gyri, with marked decreases in superior and middle frontal gyri. This profile may mirror, to some degree, the discrete pattern of accelerated gray matter loss identified here (Fig. 1). Whether or not this increased metabolism represents an adaptive or compensatory response to cell loss in superior frontal systems, a similar underlying pathophysiology may underlie these structural, metabolic, and functional impairments as the disease develops.
The early parietal deficits observed here are consistent with recent functional MRI studies in adult patients showing marked parietal activation deficits in working memory tasks (38). Recent functional imaging studies with positron emission tomography (39, 40) and functional MRI (41) also show a diminished activation of the sensorimotor cortex and supplementary motor area during motor tasks (finger-to-thumb opposition) in schizophrenia. Implication of cortical motor systems is also consistent with premorbid motor impairments, consistently noted in studies of COS. In frontal cortices, regions of the fastest progressive gray matter loss terminated anteriorly in the frontal eye fields. Visual search tasks are thought to tap a key attentional dysfunction in schizophrenia (42), namely a deficit in the ability to hone in on the most important elements in a picture and a tendency to stare instead of engaging in active visual search. By studying exploratory eye movements during scene perception, impairments have been observed in schizophrenic adolescents in the basic control of exploratory eye movements, suggesting that they stared more and had difficulty in the top-down control of selective attention and visual search. Continuous attrition of gray matter in frontal eye fields may underlie some of the deficit symptoms in visual attention. The marked anterior limit of the loss pattern around the anterior limit of the frontal eye fields (Brodmann area 8) may indicate an anatomically specific progression that has a direct impact on the systems supporting attentional dysfunction.
Recent neuropathological studies (43), evaluating regional neuronal density postmortem, have found altered cell packing of pyramidal and nonpyramidal neurons in the schizophrenic cortex, with a disproportionate reduction in layer V of prefrontal area 9. Pathologic and in vivo MRI studies jointly suggest that neuronal atrophy may be one anatomic substrate for deficient information processing in schizophrenia. Altered laminar density of cells in the schizophrenic cortex and moderate reductions in cortical thickness may be major contributors to the intense dynamic processes of gray matter loss that are imaged here in vivo and mapped as they spread from parietal to frontal and temporal regions.
Developmental Implications. Neurodevelopmental theories of the onset of schizophrenia posit disturbances, either pre- or postnatal, in the processes of neuronal migration (3, 44), or in synaptic pruning, which intensifies around the age of 5 years (15, 45-47). Our data indicate that structural changes clearly progress after psychosis onset and well into adolescence, consistent with earlier reports of ventricular expansion and overall lobar reduction (7, 9, 48). Cross-sectional studies of this COS population have also found a failure of normal maturation in neurological test performance during adolescence (49). This level of performance is also consistent with several recent brain structure studies showing more subtle but significant progressive cortical gray matter loss in adult-onset schizophrenia (8, 11, 50, 51). Perhaps surprisingly in this cohort, whereas parietal and frontal/motor deficits precede puberty, temporal deficits do not. It is probable that early neurodevelopmental abnormalities and later gray matter loss are related, as genes affecting prenatal development may also have roles in later brain maturation (45, 52, 53). Intriguingly, the earliest deficits occur in a region of parietal cortices where progressive cortical change occurs significantly in both healthy and schizophrenic subjects in the teenage years. In adults, parietal deficits appear to be mediated by environmental (nongenetic) factors, as the mathematical pattern of these deficits distinguishes schizophrenic adult twins from their healthy, genetically identical, monozygotic co-twins. Finally, the frontal and temporal territories, which were spared when our cohort was first scanned, are later engulfed by the wave of tissue loss. In these regions, deficits in adult patients appear to be highly heritable (32). By dissociating early brain structure deficits that predate psychosis onset, those that progress, and those that begin in adolescence, dynamic and genetic brain mapping may shed light on the triggers of schizophrenia. These findings are consistent with the notion that activation of some nongenetic trigger contributes to the onset and initial progression of the illness (54, 55). [emphasis added]
| |
Acknowledgements |
|---|
Grant support was provided by P41 RR13642, National Institutes of Health intramural funding, LM/MH05639, NS38753, and P20 MH/DA52176.
| |
Abbreviations |
|---|
NOS, not otherwise specified; SAPS/SANS, Scales for the Assessment of Positive and Negative Symptoms; COS, childhood-onset schizophrenia.
| |
Footnotes |
|---|
![]() To whom reprint requests should be addressed at: Laboratory of Neuro
Imaging, Department of Neurology, Reed Neurological Research
Center, Room 4238, University of California School of Medicine,
710 Westwood Plaza, Los Angeles, CA 90095-1769. E-mail:
thompson@loni.ucla.edu
.
To whom reprint requests should be addressed at: Laboratory of Neuro
Imaging, Department of Neurology, Reed Neurological Research
Center, Room 4238, University of California School of Medicine,
710 Westwood Plaza, Los Angeles, CA 90095-1769. E-mail:
thompson@loni.ucla.edu
.
This paper was submitted directly (Track II) to the PNAS office.
§ Cannon, T. D., Thompson, P. M., van Erp, T. G. M., Toga, A. W., Huttunen, M., Lönnqvist, J. & Standertskjöld-Nordenstam, C.-G., A Probabilistic Atlas of Cortical Gray Matter Changes in Monozygotic Twins Discordant for Schizophrenia, Proceedings of the International Congress on Schizophrenia Research, Whistler, Canada, April 28-May 2, 2001 (available at http://www.loni.ucla.edu/~thompson/HBM2001/ty_HBM2001.html).
| |
References |
|---|
| 1. | Jacobsen, L. K. & Rapoport, J. L. (1998) J. Child Psychol. Psychiatry 39, 101-113 |
| 2. | Feinberg, I. (1982) J. Psychiatr. Res. 17, 319-334 |
| 3. | Weinberger, D. R. (1995) in Schizophrenia, eds. Hirsch, S. R. & Weinberger, D. R. (Blackwood, London), pp. 294-323. |
| 4. | Barondes, S. H. , Alberts, B. M. , Andreasen, N. C. , Bargmann, C. , Benes, F. , Goldman-Rakic, P. , Gottesman, I. , Heinemann, S. F. , Jones, E. G. , Kirschner, M. , et al. (1997) Proc. Natl. Acad. Sci. USA 94, 1612-1614 |
| 5. | McGlashan, T. H. & Hoffman, R. E. (2000) Arch. Gen. Psychiatry 57, 637-648 |
| 6. | Selemon, L. D. & Goldman-Rakic, P. S. (1999) Biol. Psychiatry 45, 17-25 |
| 7. | Rapoport, J. L. , Giedd, J. , Kumra, S. , Jacobsen, L. , Smith, A. , Lee, P. , Nelson, J. & Hamburger, S. (1997) Arch. Gen. Psychiatry 54, 897-903 |
| 8. | DeLisi, L. E. , Sakuma, M. , Ge, S. & Kushner, M. (1998) Psychiatry Res. 84, 75-88 |
| 9. | Giedd, J. N. , Jeffries, N. O. , Blumenthal, J. , Castellanos, F. X. , Vaituzis, A. C. , Fernandez, T. , Hamburger, S. D. , Liu, H. , Nelson, J. , Bedwell, J. , et al. (1999) Biol. Psychiatry 46, 892-898 |
| 10. | Giedd, J. N. , Blumenthal, J. , Jeffries, N. O. , Castellanos, F. X. , Liu, H. , Zijdenbos, A. , Paus, T. , Evans, A. C. & Rapoport, J. L. (1999b) Nat. Neurosci. 2, 861-863 |
| 11. | Mathalon, D. H. , Sullivan, E. V. , Lim, K. O. & Pfefferbaum, A. (2001) Arch. Gen. Psychiatry 58, 148-157 |
| 12. | Yakovlev, P. I. & Lecours, A. R. (1967) in Regional Development of the Brain in Early Life, ed. Minkowski, A. (Davis, Philadelphia), pp. 3-65. |
| 13. | Jernigan, T. L. & Tallal, P. (1990) Dev. Med. Child Neurol. 32, 379-385 |
| 14. | Pfefferbaum, A. , Mathalon, D. H. , Sullivan, E. V. , Rawles, J. M. , Zipursky, R. B. & Lim, K. O. (1994) Arch. Neurol. 51, 874-887 |
| 15. | Sowell, E. R. , Thompson, P. M. , Holmes, C. J. , Jernigan, T. L. & Toga, A. W. (1999) Nat. Neurosci. 2, 859-861 |
| 16. | McCarley, R. W. , Wible, C. G. , Frumin, M. , Hirayasu, Y. , Levitt, J. J. , Fischer, I. A. & Shenton, M. E. (1999) Biol. Psychiatry 45, 1099-1119 |
| 17. | Thompson, P. M. , Mega, M. S. , Woods, R. P. , Zoumalan, C. I. , Lindshield, C. J. , Blanton, R. E. , Moussai, J. , Holmes, C. J. , Cummings, J. L. & Toga, A. W. (2001) Cereb. Cortex 11, 1-16 |
| 18. | American Psychiatric Association. (1987) Diagnostic and Statistical Manual of Mental Disorders (Am. Psychiatric Assoc., Washington, DC) revised. |
| 19. | Puig-Antich, J. , Orvaschel, H. , Tabrizi, M. A. & Chambers, W. (1980) Schedule for Affective Disorders and Schizophrenia for School-Age Children: Epidemiologic Version (New York State Psychiatric Institution, New York, and Yale Univ. School of Medicine, New Haven, CT). |
| 20. | Reich, W. , Welner, Z. & Herjanic, B. (1990)
Diagnostic Interview for Children and Adolescents Revised |
| 21. | Andreasen, N. C. (1983) Scale for the Assessment of Positive Symptoms (SAPS) and Scale for the Assessment of Negative Symptoms (SANS) (Univ. of Iowa College of Medicine, Iowa City). |
| 22. | Overall, J. E. & Gorham, D. R. (1962) Psychol. Rep. 10, 799-812 |
| 23. | Kumra, S. , Briguglio, C. , Lenane, M. , Goldhar, L. , Bedwell, J. , Venuchekov, J. , Jacobsen, L. K. & Rapoport, J. L. (1999) Am. J. Psychiatry 156, 1065-1068 |
| 24. | Woods, R. P. , Cherry, S. R. & Mazziotta, J. C. (1992) J. Comput. Assist. Tomogr. 16, 620-633 |
| 25. | Zijdenbos, A. P. & Dawant, B. M. (1994) Crit. Rev. Biomed. Eng. 22, 401-465 |
| 26. | Wright, I. C. , McGuire, P. K. , Poline, J. B. , Travere, J. M. , Murray, R. M. , Frith, C. D. , Frackowiak, R. S. & Friston, K. J. (1995) NeuroImage 2, 244-252 |
| 27. | Ashburner, J. & Friston, K. J. (2000) NeuroImage 11, 805-821 |
| 28. | Thompson, P. M. , Woods, R. P. , Mega, M. S. & Toga, A. W. (2000) Hum. Brain Mapp. 9, 81-92 |
| 29. | Thompson, P. M. , Mega, M. S. , Narr, K. L. , Sowell, E. R. , Blanton, R. E. & Toga, A. W. (2000) in SPIE Handbook on Medical Image Analysis, ed. Fitzpatrick, M. (Soc. Photo-Optical Instrumentation Engineers, Bellingham, WA), pp. 1063-1131. |
| 30. | Gogate, N. , Giedd, J. N. , Jansen, K. & Rapoport, J. L. (2001) Clin. Neurosci. Res. 1, 283-290 |
| 31. | Lawrie, S. M. & Abukmeil, S. S. (1998) Br. J. Psychiatry 172, 110-120 |
| 32. | Cannon, T. D. , Huttunen, M. O. , Lonnqvist, J. , Tuulio-Henriksson, A. , Pirkola, T. , Glahn, D. , Finkelstein, J. , Hietanen, M. , Kaprio, J. & Koskenvuo, M. (2000) Am. J. Hum. Genet. 67, 369-382 |
| 33. | Nicolson, R. , Lenane, M. , Brookner, F. , Gochman, P. , Kumra, S. , Spechler, L. , Giedd, J. N. , Thaker, G. K. , Wudarsky, M. & Rapoport, J. L. (2001) Comprehensive Psychiatry 42, 319-325 |
| 34. | Cohen, B. M. & Wan, W. (1996) Am. J. Psychiatry 153, 104-106 |
| 35. | Melis, M. , Diana, M. & Gessa, G. L. (1999) Eur. J. Pharmacol. 366, R11-R13 |
| 36. | Goldberg, T. E. & Weinberger, D. R. (1996) J. Clin. Psychiatry 57, 62-65 |
| 37. | Jacobsen, L. K. , Hamburger, S. D. , Van Horn, J. D. , Vaituzis, A. C. , McKenna, K. , Frazier, J. A. , Gordon, C. T. , Lenane, M. C. , Rapoport, J. L. & Zametkin, A. J. (1997) Psychiatry Res. 75, 131-144 |
| 38. | Menon, V. V. , Anagnoson, R. T. , Mathalon, D. H. , Glover, G. H. & Pfefferbaum, A. (2001) NeuroImage 13, 433-446 |
| 39. | Günther, W. , Brodie, J. D. , Bartlett, E. J. , Dewey, S. L. , Henn, F. A. , Volkow, N. D. , Alper, K. , Wolkin, A. , Cancro, R. & Wolf, A. P. (1994) Eur. Arch. Psychiatry Clin. Neurosci. 244, 115-125 |
| 40. | Spence, S. A. , Brooks, D. J. , Hirsch, S. R. , Liddle, P. F. , Meehan, J. & Grasby, P. M. (1997) Brain 120, Part 11, 1997-2011 |
| 41. | Schroder, J. , Essig, M. , Baudendistel, K. , Jahn, T. , Gerdsen, I. , Stockert, A. , Schad, L. R. & Knopp, M. V. (1999) NeuroImage 9, 81-87 |
| 42. | Karatekin, C. & Asarnow, R. F. (1999) J. Abnormal Child Psychol. 27, 35-49 |
| 43. | Selemon, L. D. , Rajkowska, G. & Goldman-Rakic, P. S. (1995) Arch. Gen. Psychiatry 52, 805-820. |
| 44. | Cannon, T. D. , Mednick, S. A. & Parnas, J. (1989) Arch. Gen. Psychiatry 46, 883-889 |
| 45. | Huttenlocher, P. R. (1979) Brain Res. 163, 195-205 |
| 46. | Feinberg, I. (1990) Schizophr. Bull. 16, 567-570 |
| 47. | Huttenlocher, P. R. & Dabholkar, A. S. (1997) J. Comp. Neurol. 387, 167-178 |
| 48. | Lim, K. O. , Harris, D. , Beal, M. , Hoff, A. L. , Minn, K. , Csernansky, J. G. , Faustman, W. O. , Marsh, L. , Sullivan, E. V. & Pfefferbaum, A. (1996) Biol. Psychiatry 40, 4-13 |
| 49. | Karp, B. I. , Garvey, M. , Jacobsen, L. K. , Frazier, J. A. , Hamburger, S. D. , Bedwell, J. S. & Rapoport, J. L. (2001) Am. J. Psychiatry 158, 118-122 |
| 50. | Lieberman, J. A. , Alvir, J. M. , Koreen, A. , Geisler, S. , Chakos, M. , Sheitman, B. & Woerner, M. (1996) Neuropsychopharmacology 14, 13S-21S . |
| 51. | Gur, R. E. , Cowell, P. , Turetsky, B. I. , Gallacher, F. , Cannon, T. , Bilker, W. & Gur, R. C. (1998) Arch. Gen. Psychiatry 55, 145-152 |
| 52. | Burrows, R. C. , Levitt, P. & Shors, T. J. (2000) Neuroscience 96, 825-836 |
| 53. | Mirnics, K. , Middleton, F. A. , Marquez, A. , Lewis, D. A. & Levitt, P. (2000) Neuron 28, 53-67 |
| 54. | Karlsson, H. , Bachmann, S. , Schröder, J. , McArthur, J. , Fuller Torrey, E. & Yolken, R. H. (2001) Proc. Natl. Acad. Sci. USA 98, 4634-4639 |
| 55. | Lewis, D. A. (2001) Proc. Natl. Acad. Sci. USA 98, 4293-4294 |
| 56. | Shaffer, D. , Gould, M. S. , Brasic, J. , Ambrosini, P. , Fisher, P. , Bird, H. & Aluwahlia, S. (1983) Arch. Gen. Psychiatry 40, 1228-1231 |
www.pnas.org/cgi/doi/10.1073/pnas.201243998
END OF ARTICLE FROM PNAS, September 25, 2001 | vol. 98 | no. 20 | 11650-11655
Return to Previous Page on Schizophrenia